
Mai 2024 – Ausgabe 43
Angeborene Fehlbildungen der Wirbelsäule – Teil 1


Schlüsselwörter: Spinale Fehlbildungen, spinale Dysraphien, Spina bifida, Chiari-Malformation, Tethered-Cord-Syndrom
Angeborene Anomalien der Wirbelsäule und des Rückenmarks sind primäre Differenzierungs- und Entwicklungsstörungen der embryonalen Wirbelsäulenanlage. Diese können als toxische, z. B. medikamentös bedingte Schädigungen oder im Zusammenhang mit Folsäuremangel während der Schwangerschaft, als sekundäre Fehlbildungen oder Disruptionen bezeichnet werden oder im Rahmen eines Gendefektes (primäre Fehlbildungen oder Malformationen) entstehen, wobei diese Anomalien einzeln, kombiniert oder im Rahmen von bekannten Syndromen auftreten können.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
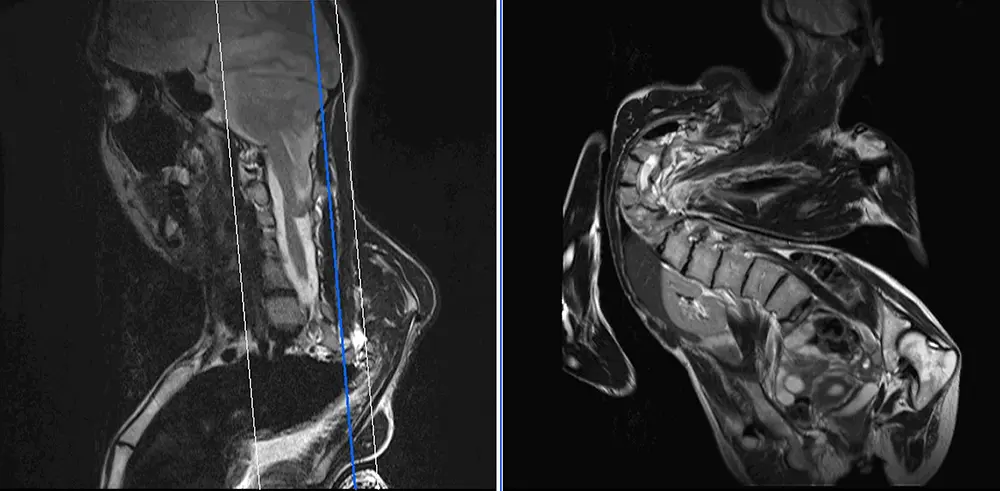
Zum Beispiel gehen angeborene Skoliosen häufig mit anderen Anomalien wie spinalen Dysraphien (20–30 %), Chiari-Malformationen (Abb. 2) und Rippenfusion einher.
Die spinalen Fehlbildungen können in Fehlbildungen knöcherner Bestandteile und/oder des Nervensystems infolge von Formationsstörungen, Segmentationsstörungen oder kombinierter Formen eingeteilt werden. In diesem ersten Teil möchten wir einen Überblick über die Entitäten, die Domäne der Neurochirurgie sind, bieten. Im zweiten Teil werden wir primär dem orthopädischen Fachgebiet zugeordnete Fehlbildungen der Wirbelsäule abhandeln.
Dysraphische Störungen
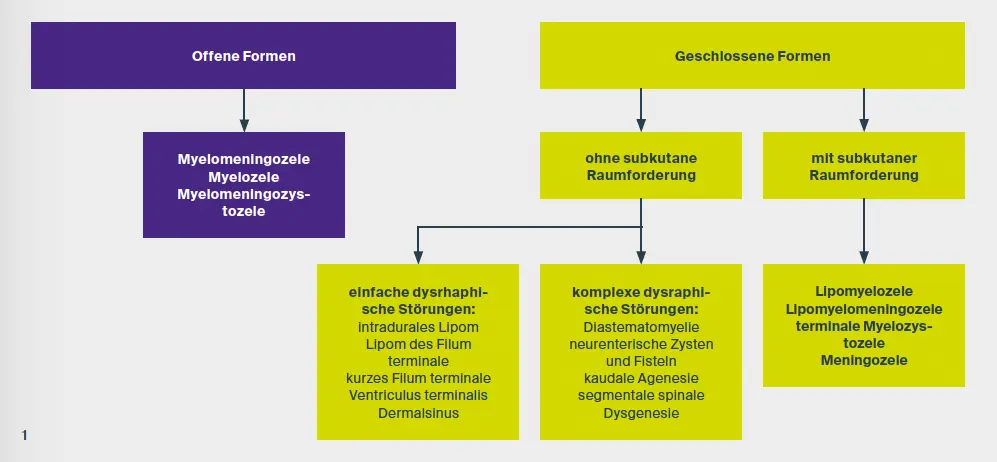
Fehlbildungen, die ihren Ursprung in einer fehlerhaften Anlage des Neuralrohres in der Embryonalzeit haben, werden als dysraphische Störungen bezeichnet. Diese werden in offene und geschlossene Formen unterteilt. Tortori-Donati et al. entwickelten auf der Basis einer Korrelation zwischen den neuroradiologischen Merkmalen der spinalen Dysraphie und klinischen Befunden eine Klassifikation, die sich in der klinischen Praxis bewährt hat (Abb. 1).
Die offene Form der dysraphischen Störungen, auch als Spina bifida aperta (völlig fehlende Hautbedeckung) oder Spina bifida cystica (schlecht epithelialisierte Hautbedeckung) bezeichnet, sind durch einen Hautdefekt mit freiliegendem Nervengewebe im Bereich der Wirbelsäule gekennzeichnet. Da das Neuralrohr sich in der Embryonalphase an verschiedenen Stellen nicht gleichzeitig schließt, wird erklärbar, warum es zu verschiedenen Entwicklungszeitpunkten zu den unterschiedlich lokalisierten und unterschiedlich ausgeprägten Störungen kommt. Frühere Schädigungen sind mit einer höheren Lokalisation und einer größeren Ausdehnung verbunden. Je später die Schädigung einsetzt, desto kaudaler liegt die Läsion. Am häufigsten kommt sie jedoch im Lendenwirbelbereich vor.
Wenn eine zystische Schwellung der Dura und der Arachnoidea durch den Spina-bifida-Defekt im Wirbelbogen hervorragt, wodurch eine flüssigkeitsgefüllte Ausbuchtung unter der Haut entsteht (das Rückenmark ist nicht verlagert), wird die Fehlbildung als Meningozele benannt. Kommt es zu einer Ausstülpung der Rückenmarkshaut und Anteilen des Rückenmarks nach außen, dann wird diese als Meningomyelozele beschrieben. Diese stellt die bedeutendste und häufigste offene Form der Spina bifida dar. Die tatsächliche schwerste, allerdings seltene Form der Spina bifida ist die Myelozele, bei der die offene Neuralplatte sekundär von Epithel bedeckt ist und sich die Neuralplatte auf der Oberfläche ausgebreitet hat.
Häufig ist die offene Spina bifida mit anderen Fehlbildungen vergesellschaftet. In der Studie von Tortori-Donati et al. hatten alle 353 Patientinnen und Patienten mit dieser Fehlbildung eine Chiari-Malformation, die häufigste Fehlbildung des kraniozervikalen Übergangs. Es werden drei Typen der Chiari-Malformation beschrieben:
- Typ I: Tiefstand der Kleinhirntonsillen in den Spinalkanal um mindestens 5 mm ohne Verlagerung der Hirnstamm-Anteile oder Erweiterung des Foramen magnum.
- Typ II: Kleinhirntonsillen, Vermis und Hirnstamm sind in den Spinalkanal verlagert. Erweiterung des Foramen magnum und Hydrozephalus sind Bestandteil dieser Form (Abb. 2). Eine Chiari-II-Malformation ist bei allen offenen dysraphischen Störungen vorhanden.
- Typ III: Hierbei sind Anteile von Kleinhirn und / oder Hirnstamm im kraniozervikalen Übergang in eine Enzephalozele herniiert.
Die geschlossene Form der dysraphischen Störung wird auch als Spina bifida occulta bezeichnet. Sie tritt bei etwa zehn Prozent der Bevölkerung auf und wird häufig zufällig entdeckt.
Bei dieser Form der Fehlbildung ist die Spaltbildung in der knöchernen Wirbelsäule mit intakter Haut bedeckt. Häufig werden hierbei, aufgrund der engen Nachbarschaft embryonaler Zellschichten, Störungen anderer Gewebetypen einbezogen, z. B. Lipome, Dermoide, Epidermoide, Teratome. Veränderungen der Haut, wegen ihres hinweisenden Charakters auch als kutane Stigmata bezeichnet, beispielsweise atypische Behaarungsmuster, Pigmentstörungen über der Fehlbildung oder kapilläre Hämangiome, werden häufig beobachtet.
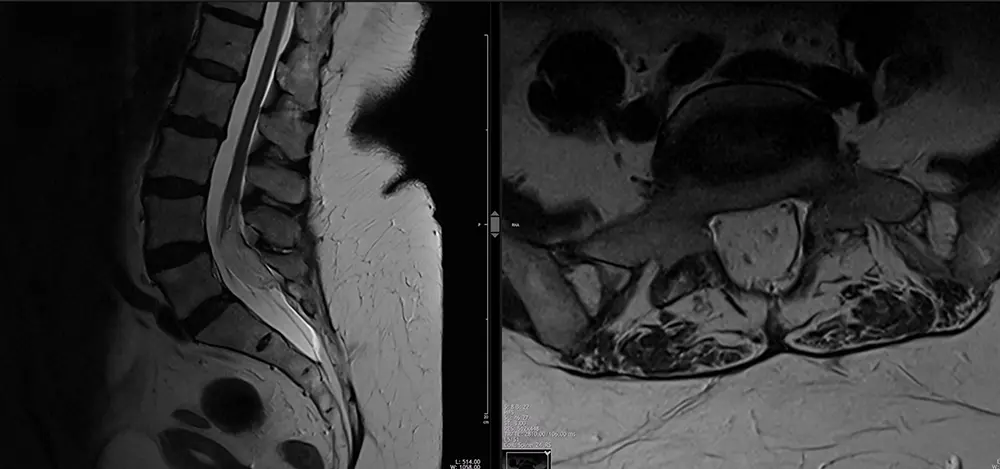
Die geschlossenen Formen der dysraphischen Störungen können mit oder ohne kutane Raumforderung einhergehen. Die spinalen Dysraphismen ohne subkutane Raumforderung können wiederum in einfache und komplexe dysraphische Zustände unterteilt werden (Abb. 1). Intradurale Lipome sind einfache dysraphische Störungen entlang der dorsalen Mittellinie und treten am häufigsten im lumbosakralen Bereich auf. Sie gehen in der Regel mit dem Tethered-Cord-Syndrom einher (Abb. 3). Dies wird als Erkrankung mit fortschreitenden neurologischen Defiziten aufgrund der Einschränkung der Beweglichkeit und Traktion des Rückenmarks definiert. Die damit verbundenen neurologischen Defizite sind schleichend und können motorischer, sensorischer, urorektaler, schmerzhafter und/oder skoliotischer Natur sein.
Das Lipom des Filum terminalis ist eine fibrolipomatöse Verdickung des Filum terminale und kann als Normvariante angesehen werden, wenn kein klinischer Hinweis auf ein Tethered-Cord-Syndrom vorliegt. Das Fortbestehen eines kleinen, mit Ependym ausgekleideten Hohlraums im Conus medullaris wird als persistierender terminaler Ventrikel bezeichnet.
Ein Dermalsinus ist eine mit Epithel ausgekleidete Fistel, die Nervengewebe oder Hirnhäute mit der Hautoberfläche verbindet.
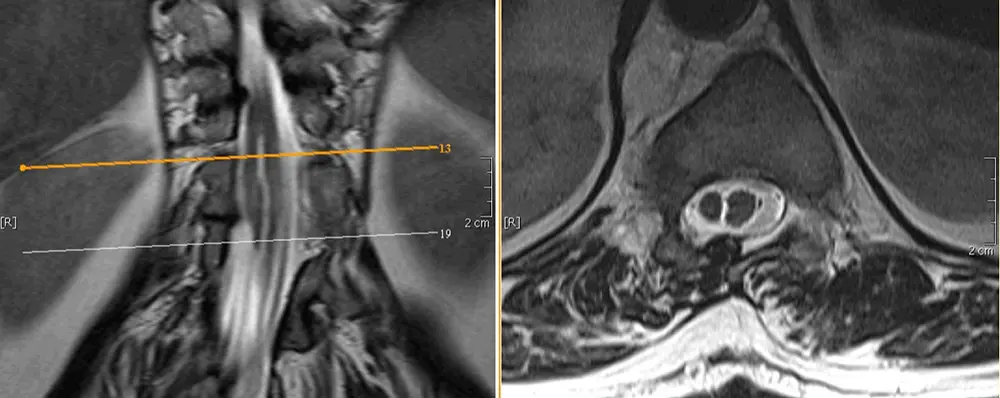
Zu den komplexen dysraphischen Störungen gehören Störungen der notochordalen Bildung (kaudale Agenesie und segmentale spinale Dysgenesie) und Störungen der notochordalen Integration in der Mittellinie (dorsale enterische Fisteln, neurenterische Zysten und Diastematomyelie). Bei der Diastematomyelie, auch als Split-Cord-Malformation bezeichnet, wird das Rückenmark durch Weichteil- ggf. durch knöchernes Gewebe in zwei Hälften geteilt und kann sich klinisch mit Skoliose und Tethered-Cord-Syndrom manifestieren. Man klassifiziert:
Split-Cord-Malformation Typ I: Die beiden Rückenmarkshälften befinden sich in getrennten Duralsäcken.
Split-Cord-Malformation Typ II: Die beiden Rückenmarkshälften befinden sich in einem gemeinsamen Duralsack.
Die geschlossene Spina bifida mit subkutaner Raumforderung ist klinisch durch das Vorhandensein einer subkutanen Fettmasse oberhalb der Gesäßfalte gekennzeichnet. Zu den Lipomen mit einem Duraldefekt zählen sowohl Lipomyelozelen, gekennzeichnet durch eine lipomatöse subkutane Masse, die in den Rückenmarkskanal eindringt und sich am fixierten Rückenmark festsetzt, als auch Lipomyelomeningozelen, gekennzeichnet durch die Kombination eines subkutanen Lipoms mit einer hinteren Meningozele.
Die Symptome bzw. die neurologischen Ausfallserscheinungen variieren abhängig vom Inhalt (nur Hirnhäute oder auch Rückenmarksgewebe), von der Ausprägung und von der Höhe des Defektes. Im Allgemeinen lassen sich dysraphische Störungen am besten mit der MRT charakterisieren, wobei in der Regel der Einsatz einer Sedierung erforderlich sein wird.
Therapie
Die Behandlung der dysraphischen Störungen sind vielfältig und können im vorliegenden Beitrag nicht detailliert ausgeführt werden. Durch eine Prophylaxe mit Folsäure und die Vermeidung von teratogenen Medikamenten in der Schwangerschaft ist die Inzidenz deutlich zurückgegangen.
Eine okkulte Spina bifida erfordert in der Regel keine Operation. Die zunehmende klinische Symptomatik im späteren Verlauf, wie z. B. Blasen-Mastdarmfunktionsstörungen und/oder neurologische Ausfälle bei Patientinnen und Patienten mit Tethered-Cord-Syndrom, kann eine Operation erforderlich machen. Bei Patientinnen und Patienten mit symptomatischen Chiari-II-Fehlbildungen ist meistens eine subokzipitale Dekompression erforderlich.
Mehrere Studien zeigen, dass Kinder mit der Diagnose einer offenen Spina bifida von einer chirurgischen Versorgung derselben noch als Fetus gegenüber der postpartalen Operation profitieren. Insofern spielt das pränatale Screening auf neurologische Anomalien eine wichtige Rolle in der frühzeitigen Diagnostik und Versorgung dieser Patientinnen und Patienten. Das Screening basiert auf routinemäßig durchgeführten Ultraschalluntersuchungen oder orientiert sich am mütterlichen Alpha-Feto-Protein (AFP)-Screening. Wird das Kind mit offener Spina bifida geboren, benötigt es wegen der hohen Infektionsgefahr eine spezielle Betreuung in einem Zentrum, in dem neonatale Verschlussoperationen rasch durchgeführt werden können.